
Research
Cell type annotation automation
Researchers working on single-cell RNA-seq, single-nucleus RNA-seq, cell type deconvolution, and spatial transcriptomics projects are frequently, at some point, tasked with cell type annotation. Specific, high-quality cell type annotations require expert domain knowledge of the tissue, and if applicable, disease. As single-cell and spatial profiling technologies improve and data generation quickens, the ability to automate high-quality cell type annotations becomes more valuable.
As a graduate student, I investigate how we can use recent advances in artificial intelligence, natural language processing, and multimodal data processing to make progress in this field.
As an undergraduate researcher, I investigated cell type annotation automation following unsupervised NMF-based clustering of bulk RNA-seq data using gene set enrichment analysis (GSEA) and related methods.
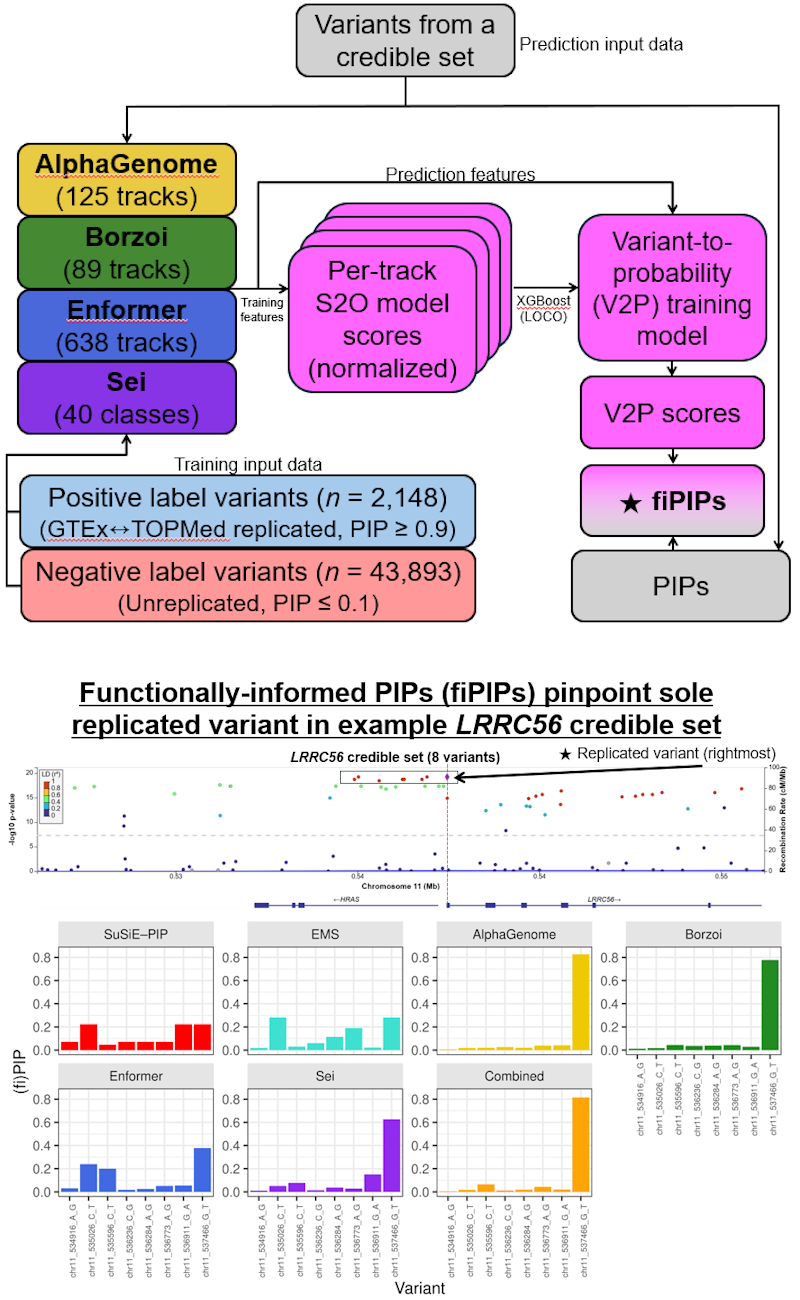
Functionally informed genetic fine-mapping
The current gold-standard methods for genetic fine-mapping are purely statistical methods that work almost entirely from association statistics and linkage disequilibrium (LD) structure. When genetic variants are in tight LD, association signals are basically indistinguishable, and identifying putatively causal variants becomes incredibly difficult.
Recent advances in artificial intelligence and deep learning have given rise to sequence-to-omics models that can predict molecular phenotypes, like RNA-seq expression, directly from DNA sequence. These models offer functional, mechanistic insight that traditional statistical methods simply can't extract from association data alone, and their implications for genetic fine-mapping and putatively causal variant identification are exciting.
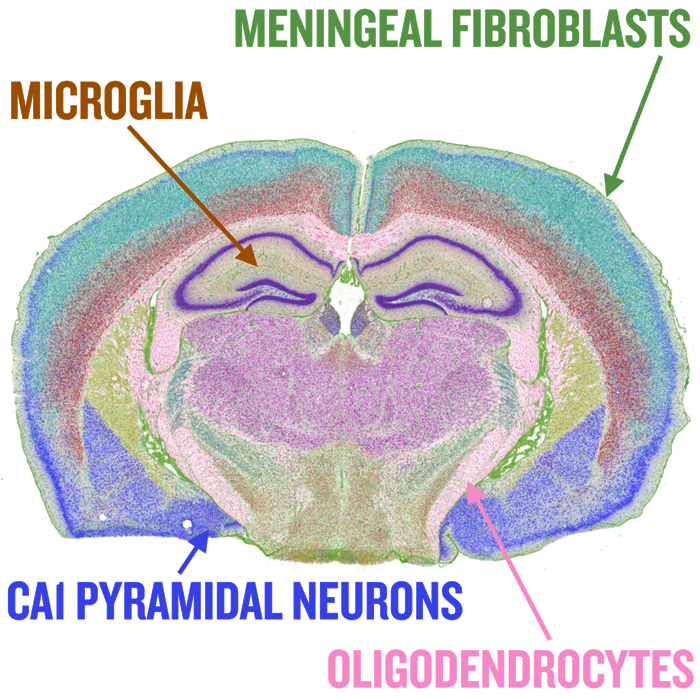
Spatial transcriptomics
I am lucky to have proximity and make contributions to the CartoScope project developed by Dr. Jun Hee Lee and Dr. Hyun Min Kang at the University of Michigan. CartoScope curates public spatial omics datasets for rapid exploration and offers high-resolution data with both morphology and cell-level information.
